
Raman spectroscopy is an analytical technique where scattered light is used to measure the vibrational energy modes of a sample. It is named after the Indian physicist C. V. Raman who was the first to observe Raman scattering (named after him) in 1928.Raman spectroscopy can provide both chemical and structural information, as well as the identification of substances through their characteristic Raman ‘fingerprint’ (spectrum). Raman spectroscopy extracts this information through the detection of Raman scattering from the sample.
Theory
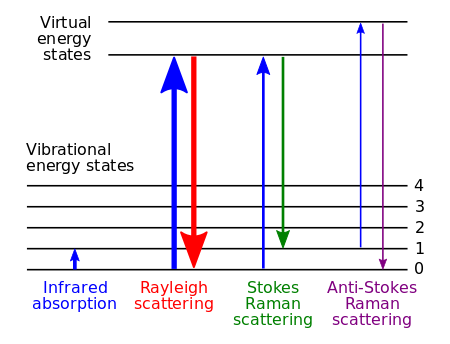
When light is scattered by molecule, the oscillating electromagnetic field of a photon induces a polarisation of the molecular electron cloud which leaves the molecule in a higher energy state with the energy of the photon transferred to the molecule. This can be considered as the formation of a very short-lived complex between the photon and molecule which is commonly called the virtual state of the molecule. The virtual state is not stable and the photon is reemitted almost immediately, as scattered light.
In the vast majority of scattering events, the energy of the molecule is unchanged after its interaction with the photon; and the energy, and therefore the wavelength, of the scattered photon is equal to that of the incident photon. This is called elastic (energy of scattering particle is conserved) or Rayleigh scattering and is the dominant process.
In a much rarer event (approximately 1 in 10 million photons)2 Raman scattering occurs, which is an inelastic scattering process with a transfer of energy between the molecule and scattered photon. If the molecule gains energy from the photon during the scattering (excited to a higher vibrational level) then the scattered photon loses energy and its wavelength increases which is called Stokes Raman scattering (after G. G. Stokes). Inversely, if the molecule loses energy by relaxing to a lower vibrational level the scattered photon gains the corresponding energy and its wavelength decreases; which is called Anti-Stokes Raman scattering. Quantum mechanically Stokes and Anti-Stokes are equally likely processes. However, with an ensemble of molecules, the majority of molecules will be in the ground vibrational level (Boltzmann distribution) and Stokes scatter is the statistically more probable process. As a result, the Stokes Raman scatter is always more intense than the anti-Stokes and for this reason, it is nearly always the Stokes Raman scatter that is measured in Raman spectroscopy.
Condition
For a molecule to exhibit a Raman effect, there must be a change in its electric dipole-electric dipole polarizability with respect to the vibrational coordinate corresponding to the rovibronic (rotational-vibrational-electronic) state. The intensity of the Raman scattering is proportional to this polarizability change. Therefore, the Raman spectrum (scattering intensity as a function of the frequency shifts) depends on the rovibronic states of the molecule.
The Raman Spectroscopy technique can be combined with other spectroscopic techniques to get additional information about the molecules. Here we will discuss the combination of Raman with Rotational Spectroscopy as well as Raman with Infra-red Spectroscopy
- Rotational Raman Spectra
Selection rule for rotational Raman spectroscopy is that the molecule must be anisotropically polarizable, which means that the distortion induced in the electron distribution in the molecule by an electric field must be dependent upon the orientation of the molecule in the field. i.e. An atom has a spherical electron distribution, and the dipole induced by an electric field of given strength is the same regardless of the orientation of the atom in that field. It is said to be isotropically polarizable.
For a hydrogen molecule, H2, the induced dipole is greater if the molecular axis is parallel to the field direction than if it is perpendicular to it. Thus a hydrogen molecule is anisotropically polarizable. Note that all linear molecules have anisotropic polarizabilities, so they may all be studied by rotational Raman spectroscopy. Many linear molecules are inactive in microwave rotational spectroscopy, so one great use of Raman spectroscopy is in the study of such molecules.
However, spherical rotors are inactive in both Raman and microwave spectroscopy (they are isotropically polarisable and have no permanent electric dipole), so may not be studied by either technique. The rotational Raman selection rules are :
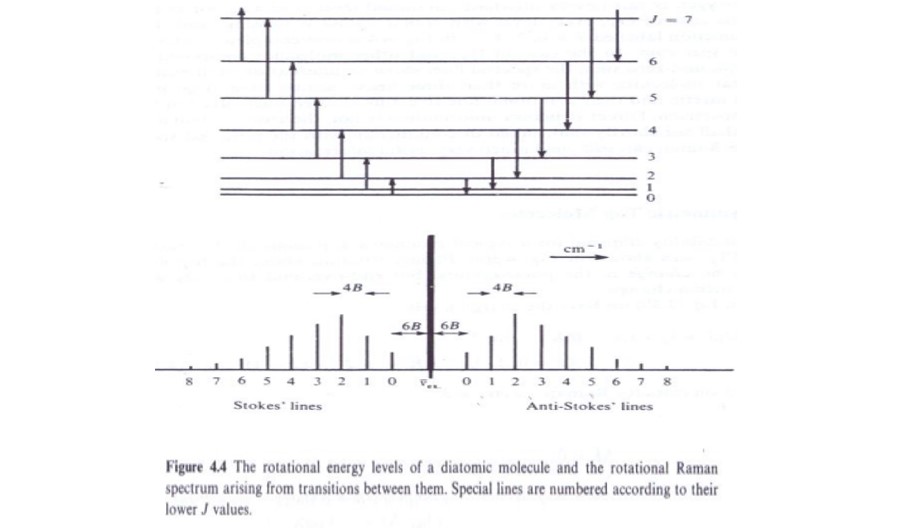
ΔJ = 0 transitions do not lead to a change in the frequency of the scattered photon, and contribute to the unshifted Rayleigh radiation that passes unaltered through the sample.
The origin of the ±2 selection rule is somewhat complex, but it should be easy to see, via a conservation of angular momentum argument, that since two photons are involved (an incoming photon that is absorbed and a scattered photon that is emitted), and each has photon has an angular momentum of one unit, the maximum change in the angular momentum of the molecule is two units.
We can apply the rotational selection rules to predict the form of the spectrum. When the molecule makes a transition with ΔJ = +2, then the interaction has imparted energy to the molecule. The scattered radiation must thus have lost energy, i.e. be at a wavenumber lower than that of the incident radiation. These transitions produce the lines in the lines in the spectrum that are known as Stokes lines: where νi is the wavenumber of the incident radiation. When the molecule makes a transition of ΔJ = -2, the incoming photon receives energy from the molecule, and thus the scattered radiation must have a higher energy, i.e. be at a higher wavenumber than the incident radiation. These transitions give rise to the anti-Stokes lines.
- Vibrational Raman Spectra
Vibrational spectroscopy is an energy sensitive method. It is based on periodic changes of dipole moments (IR) or polarizabilities (Raman) caused by molecular vibrations of molecules or groups of atoms and the combined discrete energy transitions and changes of frequencies during absorption (IR) or scattering (Raman) of electromagnetic radiation of wavelengths from 1 to 300 µm (selection rules).
One can get/detect:
• The presence of known compounds (finger print)
• The components of an unknown compound (functional groups)
• And thus a likely structure of a compound
• Changes in the concentration of a species during a reaction
• The properties of bonds (bond strength, force constants)
• State and order parameters of phase transitions
In order to describe the 3N-6 or 3N-5 different possibilities how non-linear and linear molecules containing N atoms can vibrate, the models of the harmonic and anharmonic oscillators are used. These modes of vibration (normal modes) give rise to
• Absorption bands (IR) if the sample is irradiated with polychromatic light of suitable wavelengths upon changes of the dipole moment μ = α ·E + β ·E2 + …
• Scattered light (Raman) if the sample is irradiated with monochromatic light of a suitable wavelength upon changes of the polarizabilities α with characteristic energies/frequencies/wavenumbers, intensities to be determined and analyzed.
The frequencies are in the range of 1012 to 3·1014 Hz with vibrational energies from 0.4 to 120 kJ/mole (4·10-3 – 1.24 eV), wavenumbers from 33 to 104 cm-1, and wavelengths from 300 to 1 μm.
Rule of Mutual Exclusion
For carbon dioxide, the bending and antisymmetric modes are infrared active, while the symmetric stretch mode is Raman active. This behaviour is typical of all centrosymmetric molecules. Modes that are infrared active are Raman inactive and vice versa. This is the Rule of Mutual Exclusion, which states that “no normal mode can be both infrared and Raman active in a molecule that possesses a centre of symmetry”.
3 comments for “Raman Spectroscopy – Legendshub Blog”